
Ewing Sarcoma
21.03.2025
EWING SARCOMA DIAGNOSIS AND TREATMENT
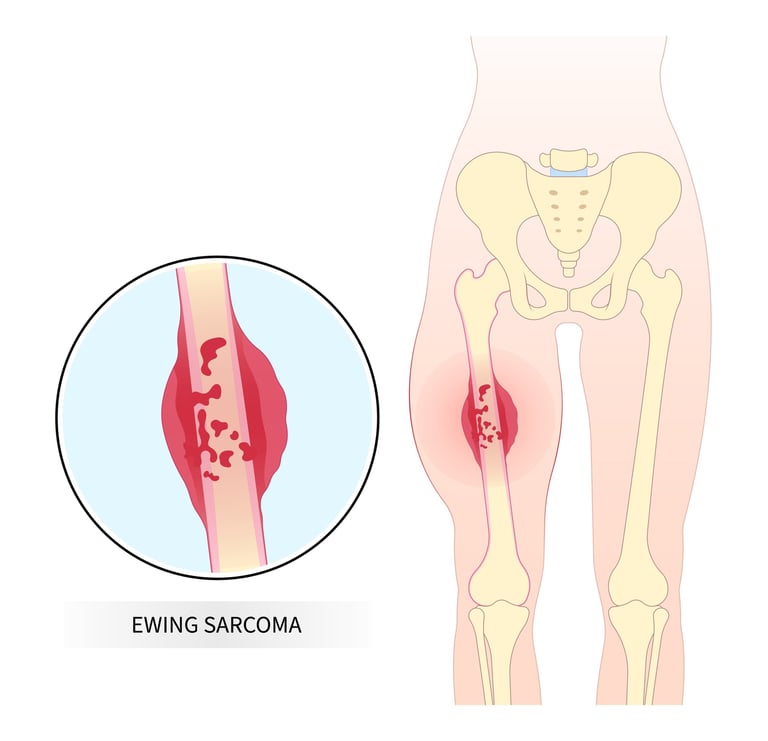
Ewing Sarcoma is an aggressive type of cancer that usually occurs during childhood and adolescence. It usually occurs in long bones, especially the legs and arms, but it can also occur in other bones such as the rib cage, spine, and pelvis. One of the most distinctive features of Ewing Sarcoma is that it can also develop in non-bone tissues. Treatment of this type of cancer usually involves a combination of surgery, chemotherapy and radiotherapy. Early diagnosis can prevent the spread of the disease and increase the chances of cure. Although Ewing Sarcoma is a rare type of cancer, it has an important place among childhood cancers and requires rapid intervention and an effective treatment plan due to its aggressive nature.
WHAT ARE THE RISK FACTORS?
Although specific risk factors for Ewing Sarcoma have not been fully determined, some factors that may be effective in the development of this type of cancer have been identified:
Age and Gender: Ewing Sarcoma usually occurs in childhood and young adulthood and is slightly more common in men than women.
Ethnicity: This type of cancer is more common in Caucasian (white-skinned) people than in other ethnic groups. It is less common in people of African and Asian descent.
Genetic Factors: Genetic mutations and chromosomal changes (e.g., t(11;22) translocation) may play a role in the development of Ewing Sarcoma. However, this cancer mostly occurs in people with no family history.
Radiation Exposure: The risk of Ewing Sarcoma may be increased in people who have previously received high doses of radiation therapy.
Genetic Diseases: Some rare genetic diseases may increase the risk of Ewing Sarcoma, but this relationship is not fully understood.
The presence of these risk factors does not necessarily mean that Ewing Sarcoma will develop. Most cases of Ewing Sarcoma occur without any obvious risk factors. Therefore, the presence or absence of risk factors alone is not a sufficient indicator for the diagnosis or treatment of the disease. In case of any symptoms or concerns, it is important to consult a physician for the most accurate information and guidance.
Image 1: Ewing sarcoma is an aggressive type of cancer that usually occurs during childhood and adolescence.


HOW IT OCCURS ?
Although the mechanisms of development of Ewing Sarcoma are complex and not fully understood, it is known that genetic mutations play an important role in the formation of this type of cancer. In the vast majority of cases of Ewing Sarcoma, a genetic change called a translocation occurs between certain chromosomes. The most common translocation occurs between chromosomes 11 and 22. As a result of this translocation, the EWSR1 gene and the FLI1 gene combine to form an abnormal fusion gene (EWS-FLI1). This fusion gene affects various pathways that control cell growth and division, causing cells to proliferate uncontrollably and develop into cancerous tumors. Additionally, these genetic changes can disrupt cell differentiation (formation) and apoptosis (programmed cell death) mechanisms, causing cells to survive and proliferate abnormally. Research is ongoing on these genetic and molecular events, as well as other factors and mechanisms that contribute to the development of Ewing Sarcoma.
WHAT ARE THE SYMPTOMS?
Signs and symptoms of Ewing Sarcoma often vary depending on where the cancer is located, but some common symptoms include:
Pain and Tenderness: Pain and tenderness in the cancerous bone area are the most common symptoms. The pain can often become constant and tend to intensify at night.
Swelling and Redness: Swelling and/or redness may occur in the area where the tumor is located. This swelling can sometimes feel warm to the touch.
Fever and Malaise: Some patients with Ewing Sarcoma may experience mild fever and general malaise.
Bone Fractures: Because the affected bone area becomes weaker, it may break more easily than usual.
Movement Restriction: If the tumor is located near the joints, it may cause limitation of movement.
Weight Loss and Loss of Appetite: Weight loss and loss of appetite may occur in association with the deterioration of general health.
Neurological Symptoms: If the tumor is near the spine, it can put pressure on nerves and cause neurological symptoms.
Each of these symptoms may indicate other health problems, so it is important to consult a physician when such symptoms are noticed. Early diagnosis of a serious condition such as Ewing Sarcoma can significantly increase treatment success.
HOW IS IT DIAGNOSED?
Diagnosing Ewing Sarcoma typically involves a series of steps and can often be challenging because its symptoms are similar to other more common bone disorders. First, the patient's medical history and physical examination are performed. After a detailed evaluation of symptoms and signs, imaging tests are performed. These tests usually include X-ray, magnetic resonance imaging (MRI), and computed tomography (CT) scans and help determine the location and size of the tumor and the tissues it affects. In case of suspicion of Ewing Sarcoma, definitive diagnosis is usually made by biopsy. During a biopsy, a small tissue sample is taken from the tumor and examined under a microscope. This examination is necessary to determine the type and characteristics of the cancer. Additionally, other tests such as a bone scan or positron emission tomography (PET) may be performed to determine the extent of the tumor. Genetic testing can be used to identify the presence of chromosomal translocations that are particularly typical of Ewing Sarcoma. This comprehensive evaluation process is critical to making an accurate diagnosis and determining the appropriate treatment plan.
WHAT ARE THE PATHOLOGICAL TYPES?
Ewing Sarcoma is part of a group of cancer types known as Ewing Sarcoma Family Tumors (ESFT). Pathological types within this group are divided according to cell type and microscopic features of the tumor. The main pathological types within the Ewing Sarcoma family are:
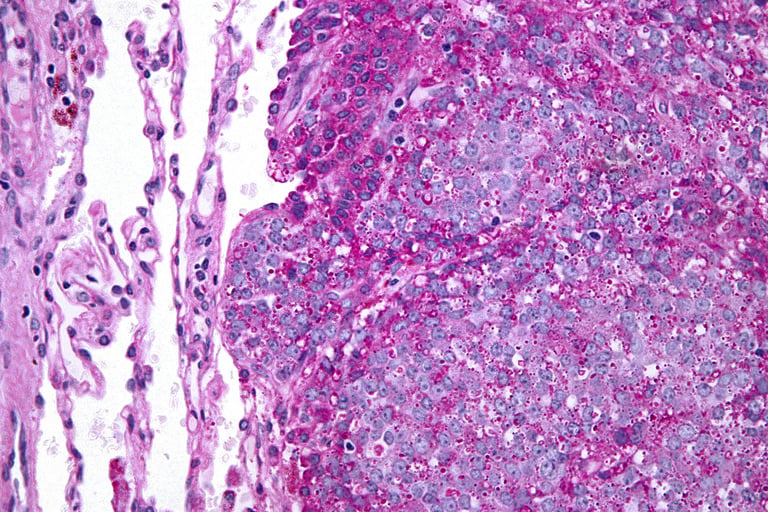
Classic Ewing Sarcoma: It is the most common type. It is characterized as small, round, blue cells under the microscope. These cells are often densely arranged and contain specific translocations.
Atypical Ewing Sarcoma: Unlike classic Ewing Sarcoma, in this type the cells may be larger and less densely arranged. These tumors, which have atypical features, may exhibit a different behavior from the classical type.
Ewing Sarcoma/PNET (Primitive Neuroectodermal Tumor): This type is considered a variant of Ewing Sarcoma and carries similar genetic features. However, PNETs display similar properties to nerve cells and may contain signs of neuroectodermal differentiation.
Extraskeletal Ewing Sarcoma: This type develops in non-bone tissues. Extraskeletal Ewing Sarcoma is a tumor located in soft tissues and generally shows similar histological features to classical Ewing Sarcoma.
Each of these pathological types is determined by microscopic examination and genetic testing. It is important to determine the pathological type of the tumor for treatment planning and prognosis (estimation of the course of the disease). Although each type requires similar treatment approaches, different treatment strategies may be required in some cases. Therefore, accurate identification of the pathological type of Ewing Sarcoma plays a critical role in the patient's treatment process.In the diagnosis and treatment of Ewing Sarcoma, it is of great importance to examine various receptors and molecular markers found on tumor cells. Some key receptors and proteins that stand out in this type of cancer, especially at the genetic and molecular level, are: CD99 is a surface protein that is generally highly expressed in Ewing Sarcoma cells and is considered one of the main markers in identifying this tumor. EWS-FLI1 Fusion Protein is an important molecular target found in most Ewing Sarcomas and resulting from chromosomal translocation. IGF-1 Receptor (Insulin-like Growth Factor 1) is another critical molecular target thought to be associated with tumor growth and spread. VEGF (Vascular Endothelial Growth Factor) is a factor that helps tumor cells nourish and grow by creating new blood vessels, and treatments directed at VEGF aim to reduce the tumor's blood flow and thus limit its growth. NKX2.2 is one of the genes regulated by the EWS-FLI1 fusion protein, is found at high levels in some Ewing Sarcomas and is seen as a potential treatment target.
Image 2: The diagnosis of Ewing sarcoma is made by pathological examination and EWS-FLI1 fusion is frequently detected.
HOW IS TUMOR STAGING DONE?
Tumor staging in Ewing Sarcoma is a comprehensive evaluation process to determine the extent of the disease and its location in the body. This process takes into account factors such as the extent of the disease, the spread of the tumor from its starting point, and the presence of metastasis (cancer spreading to other organs). Staging is vital for treatment planning and prognosis (predicting the course of the disease). Steps followed when staging the tumor in Ewing Sarcoma:
Imaging Tests: Various imaging tests are used to determine the size and location of the tumor and whether it has spread to surrounding tissues. These include X-ray, magnetic resonance imaging (MRI), computed tomography (CT), and positron emission tomography (PET) scans.
Biopsy: A tissue sample is taken from the tumor and examined under a microscope. This is necessary to determine the type and sometimes stage of the tumor.
Bone Scan: A bone scan is usually done to determine whether cancer has spread to other bones in the body.
Laboratory Tests: Laboratory tests, such as blood and urine tests, are used to evaluate general health status and detect certain tumor markers.
Ewing Sarcoma is generally divided into four main stages:
Stage I: It is not used for Ewing sarcoma. This is because all Ewing sarcomas are high grade (G2 or 3). Stage I is used for other types of bone cancer.
Stage II: It is divided into two groups.
Stage IIA. The tumor does not exceed 8 cm in diameter and is high grade. The cancer has not spread to nearby lymph nodes or organs in other parts of the body.
Stage IIB. The tumor is larger than 8 cm and is high grade. The cancer has not spread to nearby lymph nodes or organs in other parts of the body.
Stage III: It means that the tumor is in more than one spot in the same bone and is high grade. The cancer has not spread to nearby lymph nodes or organs in other parts of the body.
Stage IV: It is divided into two groups.
Stage IVA: Cancer has spread to the lungs but not to the lymph nodes or organs in other parts of the body. It can be of any size or grade.
Stage IVB: Cancer has spread to nearby lymph nodes. It may or may not have spread to organs in other parts of the body. It can be of any size or grade.
This staging process plays a fundamental role in developing a treatment plan specifically tailored to each patient's condition and influences the patient's response to treatment, survival rates, and potential treatment options.
HOW IS TREATMENT DONE ACCORDING TO STAGES?
Treatment of Ewing Sarcoma varies depending on the stage of the cancer, the patient's age, general health condition and the location of the tumor in the body. Although treatment approaches vary for each stage, a combination treatment is usually applied.
General treatment approaches according to the stages of Ewing Sarcoma:
Stage 2 (Local Disease)
Chemotherapy: The first stage of treatment is usually the use of chemotherapy to shrink the tumor and kill microscopic cancer cells in the body.
Surgery: Whenever possible, the aim is to surgically remove the entire tumor. Surgery may take the form of organ-sparing operations, depending on the location of the tumor.
Radiotherapy: If surgery is not possible or the tumor has not been completely removed, radiotherapy may be applied. It is sometimes used together with surgery.
Stage 3 (Locally Advanced Disease)
Chemotherapy: In Stage 3, chemotherapy forms the basis of treatment. Often a more aggressive chemotherapy regimen can be applied.
Surgery and/or Radiotherapy: Combinations of surgery and/or radiotherapy may be used depending on the areas where the tumor has spread.
Stage 4 (Metastatic Disease)
Chemotherapy: In stage 4, chemotherapy is the main component of treatment. As a systemic treatment, it aims to control the spread of cancer to other parts of the body.
Supportive Treatment: Additional treatments directed to the areas of metastasis (for example, radiotherapy for lung metastases) may be applied.
Surgery and Radiotherapy: It may be possible to remove the main tumor and some metastatic lesions with surgery and/or treatment with radiotherapy.
General Approach in All Stages
Supportive Care: Supportive care is important to improve the patient's quality of life and manage side effects during and after treatment.
Clinical Trials: Participation in clinical trials on new methods for treating Ewing Sarcoma may be an option for some patients.
The treatment plan is customized to each patient's unique situation and carried out by a multidisciplinary team. This team may include medical oncologists, surgeons, radiation oncologists, pathologists, pediatric specialists, and other oncology physicians. Participation of the patient and his family in the treatment process and taking an active role in decision-making processes is important in increasing the success of treatment and the general well-being of the patient.
Image 3: Ewing sarcoma can cause complaints such as pain, weight loss and fever.
WHAT ARE THE SYSTEMIC DRUGS USED IN TREATMENT?
The various medications and treatment methods used in the treatment of Ewing Sarcoma may vary depending on the extent of the cancer and the patient's general health condition. Here are the main drugs and treatment methods used in the treatment of Ewing Sarcoma:
Chemotherapy Drugs
Vincristine: It is a chemotherapy drug frequently used in cancer types such as Ewing Sarcoma.
Doxorubicin: A broad-spectrum chemotherapy agent is commonly used in the treatment of Ewing Sarcoma.
Cyclophosphamide: It may have side effects that include bone marrow suppression, but it is an effective drug in the treatment of Ewing Sarcoma.
Etoposide: Stops the growth of cancer cells by inhibiting DNA synthesis.
Ifosfamide: Often used in combination treatments and may be particularly effective for metastatic disease.
Smart Drug Therapies
Tyrosine kinase inhibitor: Inhibits the growth and proliferation of tumor cells. Ex: Regorafenib and cabozantinib.
IGF-1 Receptor Inhibitors: Target IGF-1 receptors that play a role in the growth of Ewing Sarcoma cells.
mTOR Inhibitors: Target mTOR pathways that regulate cell growth and proliferation.
Immunotherapy
The use of immunotherapy in the treatment of Ewing Sarcoma is not yet widespread and is usually evaluated as part of clinical trials.
These treatment options are often used together and/or sequentially to increase their effectiveness in treating Ewing Sarcoma. The treatment plan for each patient is customized based on their specific situation and the characteristics of the cancer. Advances in Ewing Sarcoma treatment are continually advancing through clinical research and trials. Therefore, it is important for patients and physicians to be informed about the most current treatment options and clinical studies.
HOW SHOULD FOLLOW-UP BE CARRIED OUT AFTER RECOVERY?
During the recovery period after Ewing Sarcoma treatment, regular follow-up and evaluation of patients is of great importance. This process is performed for the early detection of possible relapses (recurrence of the disease), management of long-term side effects of treatment, and monitoring of the patient's general health condition. Frequent medical check-ups are generally recommended for the first few years after treatment is completed (e.g., every three months in the first year, every six months in subsequent years). These checks use methods such as physical examination, laboratory tests and imaging tests (for example, X-ray, MRI, CT). During this period, it is important for the patient and their family to contact their doctor immediately in case of any new symptoms or concerns. In addition, regular evaluations for long-term side effects of treatment (e.g., growth and development problems, risk of secondary cancer, cardiovascular problems) and supportive treatments should be performed if necessary. Resources such as psychological counseling, social services and rehabilitation programs can be used to support the patient's psychological and social recovery. The follow-up plan for each individual recovering from Ewing Sarcoma should be customized to their specific needs, and a multidisciplinary approach should be adopted in this process.
Legal Information
The brand name ''Onkoloji Doktorum'' has been registered by the Turkish Patent and Trademark Office.
All content on this site is for informational purposes for patients receiving cancer treatment in Türkiye. Consult your doctor for treatment planning.
All content on the site is translated from Turkish. Translation errors may be present. An oncology physician should be consulted for incomprehensible information.
All articles and images used on our website are licensed and protected by DMCA. Unauthorized use or copying is prohibited.
Contact
Assoc. Prof. Dr. İzzet Doğan
Internal Medicine and Medical Oncology Specialist
Acıbadem Healthcare Group
Atakent ve Bakırköy Hospitals
Call for an Appointment: +90 444 55 44
Acıbadem Atakent Hospital
Telephone: +90 212 404 44 44
Acıbadem Bakırköy Hospital
Telephone: +90 212 414 44 44